Maintaining Regulatory Compliance in Medical Devices through ISO 13485 and 14971 - Juniper Publishers
Cancer Therapy & Oncology - Juniper Publishers

Abstract
International organization of standardization (ISO) has the responsibility of forming international standards across the globe. The technical community of international organization of standards drafts international standards as per the rules and directive. Most medical device manufacturers use ISO 13485 as the quality management system to demonstrate their ability for consistency in design, development, and safe delivery for their medical devices and related services meeting regulatory compliance and customer needs. ISO13485: 2016 replaces the 2003 version and will be mandatory for device manufacturers to transition over within three years. ISO 14971 is a standard for risk management for medical devices. FDA terms it as acceptable whereas in Europe it is mandatory
Keywords: ISO 13485:2016; ISO 14971; Medical device; Quality management system; Risk
Abbreviations: ISO: International Organization of Standardization; CAPA: Corrective and Preventive Actions; SCAR: Supplier Corrective Action Request; SWOT analysis: Strengths, Weakness, Opportunities and Threats analysis
Introduction
Medical device manufacturers have started insisting that their suppliers and service providers are ISO 13485 certified [1,2]. This gives them the confidence to demonstrate a quality management system in place for regulatory approvals and also increase confidence in the patients and other stakeholders. ISO 13485:2016 is the latest version of quality management system, as per international standards, for medical device companies that are in the process of design, development, manufacture, install and are service providers. This also includes manufacturers or suppliers of parts and components to the medical device companies. A proactive approach through QMS and risk management to prevent rather than receiving warning letters or denials, with current good manufacturing practices is required [2]. Medical device single audit programs is a logical, focused and efficient QMS audit program [3].
Necessity of using ISO 13485: 2016
To demonstrate that medical device of a device manufacturer meets or exceeds international standards in terms of regulatory and legal compliance, ISO 13485:2016 is required. It is also required to meet safety and risk mitigation requirements, manage the process with utmost efficiency and it will be mandatory to use ISO13485:2016 after three years, as up to two years [4]. ISO 13485:2013 can still be used but after three years, it has to be the 2016 version for all medical device companies to comply with. SCAR (Supplier Corrective Action Request) is an integral part of CAPA and applies to outsourced vendors (suppliers) and their control. These outsourced processes are identified in QMS and incorporate SCAR in the process.
Differences between ISO 13485: 2016 and 2003
- Incorporation of risk-based approaches beyond product realization. Risk is considered in the context of the safety and performance of the medical device and in meeting regulatory requirements.
- Increased linkage with regulatory requirements, particularly for regulatory documentation.
- Application to organizations throughout the life cycle and supply chain for medical devices.
- Harmonization of the requirements for software validation for different software applications (QMS software, process control software, software for monitoring and measurement) in different clauses of the standard.
- Emphasis on appropriate infrastructure, particularly for production of sterile medical devices, and addition of requirements for validation of sterile barrier properties.
- Additional requirements in design and development on consideration of usability, use of standards, verification and validation planning, design transfer and design records.
- Emphasis on complaint handling and reporting to regulatory authorities in accordance with regulatory requirements, and consideration of post-market surveillance.
- Planning and documenting corrective action and preventive action, and implementing corrective action without undue delay.
What is ISO 14971?
This is an ISO standard for medical devices especially for risk management. It also incorporated in-vitro devices (IVD). EN ISO 14971:2012 is the harmonized, current version in European Union with focus on hazards and hazardous situations, control and effectiveness of these risks, and also looks at overall residual risks [5]. Hazardous medical device would be any device that could cause potential harm to the patient and other users, like a sharp edge, radiation or a virus or bacteria. A hazard traceability matrix or risk analysis is required for risk management. The 2012 version applies only to European market devices and for all other countries ISO 14971:2007 still holds well [6,7].
The three versions of ISO 14971: 2012, 2009 and 2007- Differences
In all these versions the normative text with the requirements are the same. The difference lies in Annex ZA. ZB and ZC of 2012 version indicating to content deviations of the European directives and essential requirements (ERs) of each directive are met. An assessment of risk/ benefit analysis may effect on the design and manufacturing of medical devices and not even negligible risks are ignored and reduced as much as possible.
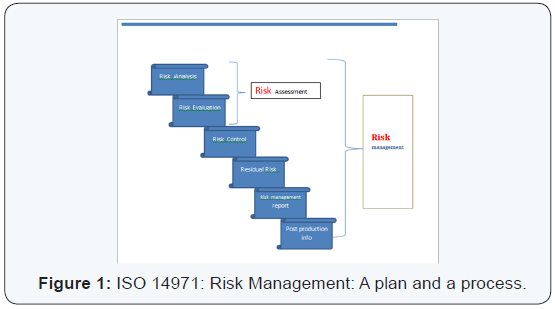
Horizontal standards:ISO 13485 and 14971: The two standards of QMS (ISO 13485) and risk management (ISO 14971) are prime examples of horizontal standards that are essential, basic requirements for general safety and performance of all the medical devices. This is in contrast to semi horizontal (ISO 11135) and vertical standards (ISO 15197), in terms of family of similar products and specific products or processes respectively. A SWOT analysis (Strengths, Weakness, Opportunities and Threat) can help to delineate risks better [8]. The warning letters from FDA are on a rise [9] (Figures 1 & 2).

Conclusion
Negative risks are avoided by medical device industry because they are not the opportunities created through positive risks that may be advantageous to these medical device companies. There has been a steady increase in the FDA warning letters. Adopting an effective QMS towards managing and streamlining issues and spending less time and resources on corrective actions, will undoubtedly help a medical device manufacturer and supplier throughout the entire life cycle of the medical device with the quality standards and risk mitigation.
To
Know More About Cancer
Therapy & Oncology International Journal Please click on:
https://juniperpublishers.com/ctoij/index.php
For
more Open Access Journals in Juniper
Publishers please click on:
https://juniperpublishers.com/index.php
For
more about Juniper
Publishers Please click on:
https://juniperpublishers0.wixsite.com/juniperpublishers


Comments
Post a Comment